
A team from Baylor College of Medicine and the Duncan Neurological Research Institute in Texas has just proposed in the journal Science Translational Medicine a new avenue for treating Rett syndrome. This strategy does not replace current care and remains tested only in the laboratory, but it directly targets the key gene of the disease, MECP2, with the idea of finely restoring its function in the brain.
Rett syndrome: the gene
MECP2 becomes a therapeutic target
THE
Rett syndrome is caused by loss-of-function mutations of the gene MECP2conductor of numerous genes necessary for the proper functioning of neurons. When the MeCP2 protein is missing, or functions poorly, neuronal networks become disorganized. Some disease-causing mutant MeCP2 proteins are less abundant and/or have reduced affinity for DNA, an essential function of this protein.
For families, this pathology remains one of the most distressing for pediatric neurologists. “Rett syndrome is a rare genetic neurodevelopmental condition that causes developmental regression, usually after 6 to 18 months of normal growth, leading to severe impairments in motor skills, speech, and communication. The disorder mainly affects girls; approximately 1 in 10,000 births“, explained Dr. Huda Zoghbi, professor emeritus at Baylor University, director of the Duncan NRI and researcher at the Howard Hughes Medical Institute. Hence the interest in an approach that attacks the gene in question rather than just the symptoms.
In mice, reintroducing a normal version of MeCP2 into the brain has already reversed symptoms and improved survival. “This is important because approximately 65% of Rett syndrome patients have partially functional MeCP2 protein that exhibits either decreased DNA binding or lower than normal abundance. Working with mouse models and cells derived from patients with Rett syndrome, our study provides proof of concept that increasing the levels of mutant MeCP2 in patients with this condition could provide therapeutic benefit.“, detailed Harini Tirumala, doctoral student in Huda Zoghbi’s laboratory.
Force switch from MeCP2-E2 to MeCP2-E1 to increase protein
The challenge is to correct this deficiency without going into excess, because too much MeCP2 causes another serious disorder, MECP2 duplication syndrome. Striking this delicate balance makes it difficult to develop safe and effective treatments.
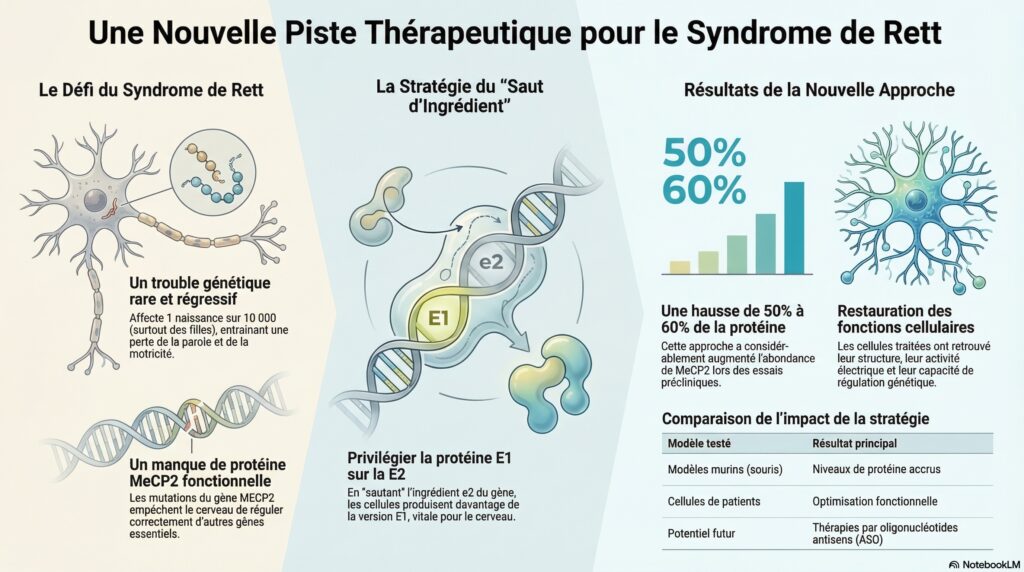
The researchers exploited another specificity of
MECP2 : This gene produces two forms of the protein, MeCP2-E1 and MeCP2-E2. E1, the majority, does not use exon e2, while E2 includes it, and no Rett mutation was found on E2.
“In summary, we knew that MeCP2-E2 differs from MeCP2-E1 in only one gene element, is less abundant than E1, is not associated with Rett syndrome, and is not required for MeCP2 function in the brain.”said Harini Tirumala. “This led us to hypothesize that directing brain cells to eliminate the e2 element would promote the production of more MeCP2-E1 protein in patients with Rett syndrome and improve the course of the disease. We tested this hypothesis in mice and on cells derived from patients with Rett syndrome.“.
 © NotebookLM
© NotebookLM
By deleting this exon 2 in mice, the team obtained a 50 to 60% increase in MeCP2 without side effects. In patient-derived neurons, the same approach restored structure, electrical activity and gene regulation.
Towards antisense oligonucleotides targeting MECP2?
To test a drug option, the researchers used morpholinos, molecules which block access to exon e2 and increase MeCP2-E1 in mice. This allowed an increase in the expression of MeCP2 and a functional improvement, but these compounds have too high a toxicity. They are now banking on antisense oligonucleotidesalready used in other genetic diseases, to reproduce this mechanism more safely.